Distrofia muscolare di Duchenne: che cos’è, cause genetiche e quadro clinico
Redazione IKOSECM
Articolo di approfondimento al corso Ecm C0604.
La distrofia muscolare di Duchenne è la più frequente fra le miopatie geneticamente determinate dell’età evolutiva e, per gravità e impatto sulla vita del paziente, una delle più impegnative da seguire. Comprenderne la base genetica, il meccanismo con cui il muscolo si deteriora e la sequenza con cui la malattia evolve è il presupposto di qualunque progetto di cura: la valutazione funzionale, la prevenzione dei danni secondari e il lavoro dell’equipe poggiano tutti su questo quadro. Questo approfondimento ricostruisce, sulla traccia di un corso ECM dedicato, che cos’è la distrofia di Duchenne, perché si manifesta, come si riconosce all’esordio e quale decorso descrive la sua storia naturale.
Questo articolo accompagna il corso FAD “La distrofia muscolare di Duchenne. Approccio fisioterapico riabilitativo”, a cura del Dott. Ivan Di Francescantonio, che eroga 10 crediti ECM. È rivolto a un’equipe ampia: medico chirurgo, fisioterapista, terapista occupazionale, terapista della neuro e psicomotricità dell’età evolutiva, infermiere e infermiere pediatrico, logopedista, psicologo, educatore professionale, podologo, tecnico ortopedico e altre figure della riabilitazione. Questa pagina ha finalità informativa e inquadra la malattia; gli aspetti valutativi e riabilitativi sono sviluppati nel percorso formativo.
Che cos'è la distrofia muscolare di Duchenne
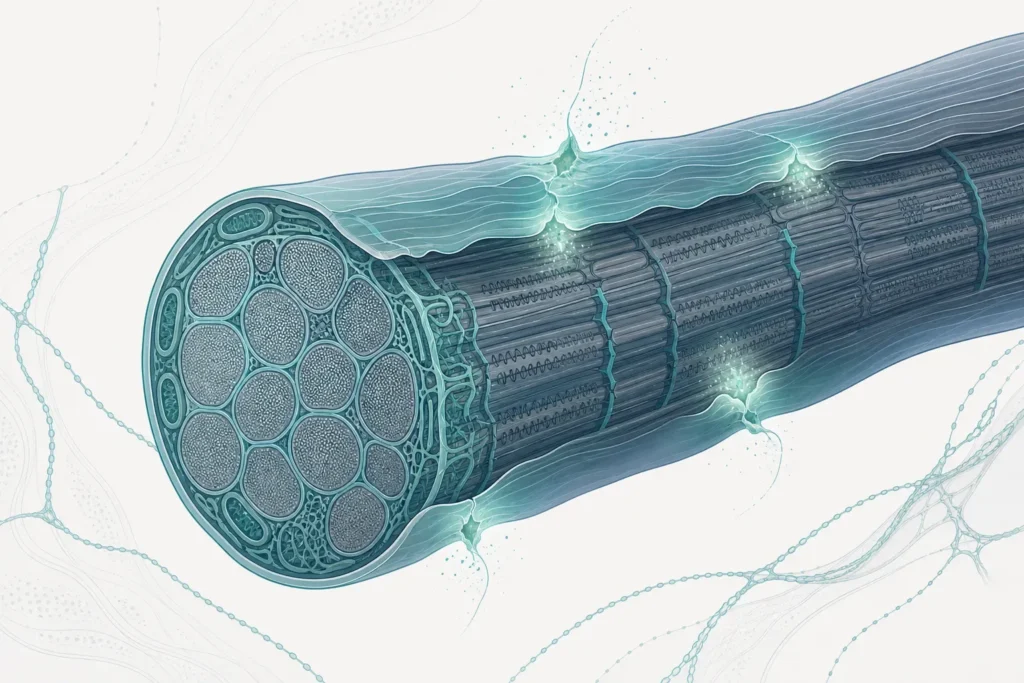
La distrofia muscolare di Duchenne (DMD) appartiene al gruppo delle distrofinopatie, le malattie legate a un difetto della distrofina. Si tratta di una miopatia strutturale, cioè di una malattia primitiva del muscolo, a carattere progressivo e degenerativo: nel tempo le fibre muscolari si deteriorano e vengono in parte sostituite da tessuto connettivo e adiposo. Non è quindi un problema nervoso periferico o di trasmissione neuromuscolare, ma una sofferenza intrinseca del muscolo stesso, che interessa il muscolo scheletrico e può coinvolgere anche quello cardiaco.
Definire con precisione di che cosa si parla è il primo passo, perché la parola “distrofia” raccoglie quadri molto diversi tra loro. La caratteristica che accomuna le distrofinopatie è il bersaglio molecolare: tutte dipendono, in misura diversa, da un’alterazione della distrofina. Ciò che cambia da una forma all’altra è quanta distrofina il muscolo riesce ancora a produrre, e questo si traduce in gravità e velocità di progressione differenti.
All’interno delle distrofinopatie il materiale del corso distingue la forma di Duchenne, la più grave, dalla forma di Becker, più lieve e a decorso più lento, accennando anche alla distrofia di Emery-Dreifuss. La differenza dipende dal tipo di alterazione del gene: nella Duchenne la mutazione interrompe del tutto la produzione di distrofina, nella Becker ne consente una versione ridotta o parzialmente funzionante. È per questo che malattie con la stessa origine genetica possono avere espressioni cliniche molto diverse: capire dove si colloca un paziente lungo questo spettro orienta le aspettative sul decorso e il progetto di presa in carico.
La genetica della DMD: trasmissione X-linked e ruolo della distrofina
Il gene coinvolto è quello della distrofina, localizzato sul cromosoma X. La trasmissione è X-linked recessiva: la malattia colpisce quasi esclusivamente i maschi, mentre le femmine sono in genere portatrici sane, perché il secondo cromosoma X compensa il gene alterato. Questo spiega perché la Duchenne sia, nella pratica, una patologia del sesso maschile, con possibilità di trasmissione attraverso le portatrici nelle generazioni successive. La conoscenza di questo meccanismo è rilevante anche sul piano familiare, perché apre il tema del consiglio genetico per i fratelli e per le portatrici.
La distrofina è una proteina del citoscheletro che ha un ruolo meccanico essenziale: stabilizza la membrana della fibra muscolare, il sarcolemma, durante le contrazioni. Funziona, in modo semplificato, come un ancoraggio che tiene insieme l’apparato contrattile interno e la parete della cellula, distribuendo le tensioni generate dal movimento. In sua assenza la membrana diventa fragile e si lesiona ripetutamente con il lavoro muscolare: ogni contrazione, che in un muscolo sano è un gesto fisiologico, diventa una micro-aggressione per la fibra priva di distrofina.
Il materiale descrive un meccanismo patogenetico in due fasi: una prima fase di necrosi e rigenerazione, in cui le fibre danneggiate tentano di ripararsi, e una fase successiva in cui prevalgono fibrosi e perdita muscolare, con infiammazione cronica e debolezza progressiva. È il passaggio dalla riparazione alla sostituzione del muscolo con tessuto non contrattile a determinare l’aggravamento clinico. Questa lettura in due tempi aiuta a comprendere perché la malattia non sia statica ma progredisca, e perché molti interventi mirino a contenere i danni che si sommano a quello primario.
Quadro clinico ed esordio: dai segni precoci alla storia naturale
I primi segni compaiono nella prima infanzia. Il materiale colloca l’esordio tipicamente fra i 3 e i 5 anni, con la maggior parte dei casi riconosciuti prima dei 4 anni. La debolezza interessa per prima la muscolatura prossimale del cingolo pelvico, e da qui la malattia progredisce in modo simmetrico verso il cingolo scapolare e i muscoli più distali e respiratori. Questa progressione, che parte dal centro del corpo per estendersi verso la periferia, è una delle chiavi per leggere il quadro: i gesti che richiedono i grandi muscoli dell’anca e delle cosce sono i primi a diventare difficili. Sono caratteristici l’andatura anserina (un cammino dondolante, “a papera”, dovuto al deficit dei muscoli dell’anca), le cadute frequenti e la difficoltà a correre, saltare e salire le scale.
Due segni semeiologici sono particolarmente evocativi. La pseudoipertrofia è un apparente aumento di volume di alcuni muscoli, soprattutto i polpacci (il tricipite surale), che assumono una consistenza gommosa: il muscolo sembra più grosso ma è in realtà infiltrato da tessuto fibroso e adiposo. Il segno di Gowers descrive il modo con cui il bambino si alza da terra arrampicandosi su se stesso, appoggiando le mani sulle gambe e risalendo lungo le cosce, per supplire alla debolezza dei muscoli estensori dell’anca.
La storia naturale descritta nel materiale è progressiva: alla perdita della deambulazione autonoma, collocata indicativamente fra i 7 e i 14 anni, seguono nel tempo le problematiche cardio-respiratorie che condizionano la prognosi, insieme a un coinvolgimento cardiaco sotto forma di cardiomiopatia dilatativa. Il materiale segnala inoltre la possibilità di un interessamento cognitivo in una parte dei pazienti. Sono dati di prognosi e percentuali che, trattandosi di una malattia rara e grave, vanno sempre contestualizzati nel singolo caso e affidati alla valutazione specialistica.
Come si arriva alla diagnosi e perché serve la valutazione funzionale
Il sospetto nasce dal quadro clinico, ma la conferma richiede esami mirati. Il materiale richiama il riscontro di valori molto elevati di creatinchinasi nel sangue (iperCKemia), espressione del danno della membrana muscolare: la creatinchinasi è un enzima che fuoriesce dalle fibre quando il sarcolemma si lesiona, e i suoi valori risultano marcatamente aumentati anche prima che il quadro clinico sia conclamato. Accanto a questo, la biopsia muscolare e soprattutto l’esame genetico permettono di identificare la mutazione del gene della distrofina, di distinguere la forma di Duchenne da quella di Becker e di orientare il consiglio genetico in famiglia.
Una volta posta la diagnosi, il percorso di cura ha bisogno di strumenti per misurare nel tempo lo stato del paziente. Il corso descrive la valutazione funzionale come quel resoconto riproducibile e obiettivo che permette di seguire l’evoluzione e calibrare gli interventi: l’esame muscolare con la scala MRC (Medical Research Council), che gradua la forza del singolo muscolo da 0 a 5, le scale funzionali (Siegel, Demos) e la misurazione goniometrica delle escursioni articolari (ROM). L’obiettivo non è una valutazione una tantum, ma il monitoraggio: confrontare misure ottenute con lo stesso metodo in momenti diversi consente di cogliere la direzione e la velocità del cambiamento.
Per questo, secondo il materiale, le valutazioni di forza e di escursione articolare vanno ripetute a intervalli regolari, più ravvicinati nelle forme a evoluzione rapida e più dilazionati nelle altre. È su questa fotografia oggettiva e aggiornata che si fondano poi le scelte assistenziali e gli aspetti riabilitativi sviluppati nel percorso formativo, che da qui prendono le mosse.
La storia naturale: come evolve la malattia nel tempo
Conoscere la traiettoria attesa della malattia è importante quanto riconoscerne i segni iniziali, perché orienta la presa in carico e l’organizzazione dell’equipe nel tempo. Il materiale descrive una storia naturale che procede per tappe abbastanza riconoscibili. Dopo l’esordio nei primi anni di vita, la debolezza progredisce dal cingolo pelvico verso quello scapolare, fino a interessare i muscoli respiratori. La perdita della deambulazione autonoma rappresenta uno snodo clinico e assistenziale rilevante, collocato dal materiale, in modo indicativo, nell’arco dell’età evolutiva.
Con il progredire della malattia entrano in primo piano le problematiche cardio-respiratorie: la riduzione della funzione ventilatoria e il coinvolgimento del muscolo cardiaco, descritto nel materiale sotto forma di cardiomiopatia dilatativa, diventano i fattori che maggiormente condizionano la prognosi. Il materiale segnala inoltre la possibilità di un interessamento cognitivo in una parte dei pazienti. È un quadro complesso, che spiega perché la Duchenne richieda una presa in carico multidisciplinare e non solo muscolo-scheletrica.
Va sottolineato un punto di metodo: i dati di prognosi, le fasce d’età e le percentuali riportate provengono dal materiale didattico del corso e descrivono un andamento generale, non il destino del singolo paziente. La variabilità individuale è ampia e la valutazione del caso resta di competenza specialistica. Per questo, in un contenuto che parla di una malattia rara e grave, è doveroso presentare questi elementi come cornice formativa e non come previsione clinica.
Domande frequenti
Che cos'è la distrofia muscolare di Duchenne?
È una miopatia strutturale geneticamente determinata, cioè una malattia primitiva del muscolo a carattere progressivo e degenerativo. Appartiene alle distrofinopatie e si trasmette con modalità X-linked. Il deterioramento delle fibre muscolari porta a una debolezza che evolve nel tempo, interessando dapprima la muscolatura dei cingoli e, in seguito, anche la funzione respiratoria e cardiaca.
Che cos'è la distrofina e qual è il suo ruolo nel muscolo?
La distrofina è una proteina del citoscheletro che stabilizza la membrana della fibra muscolare durante le contrazioni. La sua mancanza rende le fibre meccanicamente fragili: si lesionano con il lavoro muscolare, vanno incontro a cicli di necrosi e rigenerazione e, col tempo, a fibrosi e perdita di tessuto contrattile. È proprio l’assenza di distrofina a innescare il deterioramento muscolare progressivo che caratterizza la malattia.
Come si trasmette la distrofia di Duchenne e perché colpisce soprattutto i maschi?
La trasmissione è X-linked recessiva: il gene della distrofina si trova sul cromosoma X. I maschi, che hanno un solo cromosoma X, sviluppano la malattia se quel gene è alterato; le femmine, con due cromosomi X, sono in genere portatrici sane perché il secondo X compensa. Per questo la Duchenne è nella pratica una patologia del sesso maschile, trasmessa attraverso le portatrici.
Qual è la differenza tra distrofia di Duchenne e distrofia di Becker?
Entrambe sono distrofinopatie, cioè dipendono da un difetto della distrofina, ma differiscono per gravità. Secondo il materiale, nella Duchenne la mutazione interrompe del tutto la produzione di distrofina e il decorso è più grave; nella Becker il gene consente una distrofina ridotta o parzialmente funzionante, con un quadro più lieve e a evoluzione più lenta. È il tipo di alterazione genetica a spiegare la diversa espressione clinica.
Qual è l'incidenza della distrofia muscolare di Duchenne?
Il materiale del corso riporta un’incidenza di circa un caso ogni 3.500 maschi nati vivi, dato che colloca la Duchenne fra le malattie rare ma fra le più frequenti tra le distrofie muscolari dell’età evolutiva. Trattandosi di un dato epidemiologico, va sempre interpretato nel contesto delle fonti aggiornate e della popolazione di riferimento.
Quali muscoli vengono colpiti per primi nella DMD?
I primi muscoli interessati sono quelli prossimali del cingolo pelvico. Da qui la malattia progredisce in modo simmetrico verso il cingolo scapolare e, successivamente, verso i muscoli respiratori e più distali. Questa sequenza spiega perché i segni iniziali riguardino soprattutto il cammino, l’alzarsi da terra e il salire le scale, attività che richiedono la forza dei muscoli dell’anca e delle cosce.
Quali capacità motorie vengono perse per prime nella DMD?
Secondo il materiale, le prime attività a venire meno sono quelle in cui tutto il corpo si muove contro gravità, come correre e saltare, perché richiedono la maggiore quantità di lavoro muscolare. Solo in seguito diventano difficili attività progressivamente meno impegnative, come salire le scale, alzarsi da terra e da una sedia, fino al cammino. Questo ordine riflette il carico muscolare richiesto da ciascun gesto.
Che cos'è la pseudoipertrofia nella distrofia di Duchenne?
È un apparente aumento di volume di alcuni muscoli, in particolare il gastrocnemio del polpaccio, che assumono una consistenza gommosa. Il muscolo sembra più sviluppato, ma in realtà il tessuto contrattile è in parte sostituito da tessuto fibroso e adiposo. Con il progredire della malattia, segnala il materiale, la pseudoipertrofia tende a ridursi fino a scomparire.
Che cos'è il segno di Gowers?
È la modalità tipica con cui il bambino con distrofia di Duchenne si alza da terra: si arrampica su se stesso, appoggiando le mani sulle gambe e risalendo lungo le cosce, per compensare la debolezza dei muscoli estensori dell’anca. È un segno clinico evocativo, spesso fra i primi a essere notati, e può orientare verso il sospetto diagnostico.
Che cos'è l'andatura anserina?
L’andatura anserina è un cammino dondolante, definito anche “a papera”, che compare quando la debolezza interessa i muscoli stabilizzatori dell’anca. Il bacino oscilla lateralmente a ogni passo per compensare il deficit muscolare. Nella distrofia di Duchenne è uno dei segni clinici precoci, insieme alle cadute frequenti e alla difficoltà a correre e a salire le scale.
Come si diagnostica la distrofia muscolare di Duchenne?
Il percorso parte dal sospetto clinico e si conferma con esami mirati: il riscontro di valori molto elevati di creatinchinasi nel sangue (iperCKemia), la biopsia muscolare e, soprattutto, l’esame genetico, che individua la mutazione del gene della distrofina. L’analisi genetica consente anche di distinguere la forma di Duchenne da quella di Becker e di impostare il consiglio genetico in famiglia.
A che cosa serve la scala MRC nella valutazione del paziente?
La scala MRC (Medical Research Council) gradua la forza del singolo muscolo su sei livelli, da 0 (nessuna contrazione) a 5 (forza normale). Permette un resoconto riproducibile e obiettivo della forza muscolare e, ripetuta nel tempo, di seguire la progressione della malattia e di calibrare gli interventi. È uno degli strumenti della valutazione funzionale insieme alle scale di Siegel e Demos e alla misurazione goniometrica delle articolazioni.
La distrofia di Duchenne colpisce solo i muscoli?
No: pur essendo una malattia primitiva del muscolo, nel tempo coinvolge anche il muscolo cardiaco, con possibile cardiomiopatia dilatativa, e la funzione respiratoria, due aspetti che secondo il materiale condizionano la prognosi. Il materiale segnala inoltre la possibilità di un interessamento cognitivo in una parte dei pazienti. Per questo la presa in carico è multidisciplinare e coinvolge un’equipe ampia.
In conclusione
Inquadrare correttamente la distrofia muscolare di Duchenne significa tenere insieme tre piani: la base genetica (il deficit di distrofina trasmesso per via X-linked), il meccanismo con cui il muscolo si deteriora e la storia naturale che ne deriva, dai segni precoci come l’andatura anserina e il segno di Gowers fino al coinvolgimento cardio-respiratorio. È questa cornice patologica a dare senso alla valutazione funzionale e a tutto il lavoro dell’equipe. Il corso ECM “La distrofia muscolare di Duchenne. Approccio fisioterapico riabilitativo”, a cura del Dott. Ivan Di Francescantonio, sviluppa questo inquadramento e ne mostra le ricadute valutative e riabilitative, erogando 10 crediti ECM in modalità FAD per le numerose professioni dell’equipe che segue il paziente.